Мукополисахаридоз – генетическая патология

Чаще всего при таком нарушении поражается скелет и задерживается физическое развитие, а некоторые разновидности патологии проявляются отсталостью со стороны интеллектуальной сферы. Помимо этого, наблюдаются сбои в работе сердечно-сосудистой, желудочно-кишечной и нервной систем, зрительного аппарата и кожи.
Лечение мукополисахаридозов симптоматическое, прогноз в целом неблагоприятный – нарушения прогрессируют, потому что глюкозаминогликаны накапливаются. Пациенты умирают достаточно рано.
Оглавление: 1. Общие данные 2. Мукополисахаридоз типа IH 3. Мукополисахаридоз типа I-S 4. Мукополисахаридоз типа II 5. Мукополисахаридоз типа III 6. Мукополисахаридоз типа IV 7. Мукополисахаридоз типа VI 8. Мукополисахаридозы типа VII и VIII 9. Диагностика 10. Дифференциальная диагностика 11. Осложнения 12. Лечение 13. Профилактика 14. Прогноз
Общие данные
Мукополисахаридоз изучили относительно недавно – в 20 столетии. Сперва он расценивался как одна патология, затем в силу разнообразия стали выделять его типы. Объединяющим фактором всех мукополисахаридозов является то, что в органах и тканях скапливаются кислые мукополисахариды, а причиной такого нарушения является неполноценность некоторых клеточных ферментов, которая заложена в генотипе и передается по наследству.
Лечение пациентов с мукополисахаридозом проводят ортопеды, но при этом также требуются клинические усилия кардиологов, офтальмологов, неврологов, отоларингологов и некоторых других специалистов.
Мукополисахаридоз типа IH
Мукополисахаридоз типа IH еще называют синдромом Гурлера. Он диагностируется у 1 новорожденного на 20-25 тысяч.
Первые признаки заболевания начинают появляться в течение первого года жизни ребенка, но полная клиническая картина может развиться только к 2 годам.
Наиболее типичными симптомами болезни являются:
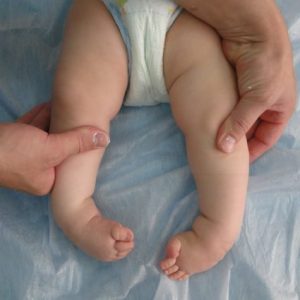
- деформация черепа – он становится похож на лодочный киль. Такое состояние называется скароцефалией;
- дыхание осуществляется через рот, так как носовое дыхание нарушено из-за увеличения аденоидов, а также аномалий внутриутробного развития в области лицевого черепа;
- деформация конечностей и некоторых других фрагментов скелета;
- отставание в росте;
- искривление позвоночника (симптом «кошачьей спины» в сидячем положении);
- укорочение шеи;
- нетипичное высокое расположение лопаток;
- выпирание вперед нижних ребер;
- увеличение ширины кистей – они при этом напоминают лапу с когтями;
- развитие контрактур (снижения объема движений) с постепенным втягиванием в патологический процесс разных суставов. Сперва страдают локтевые и плечевые суставы, далее процесс распространяется на коленные, тазобедренные и голеностопные сочленения.
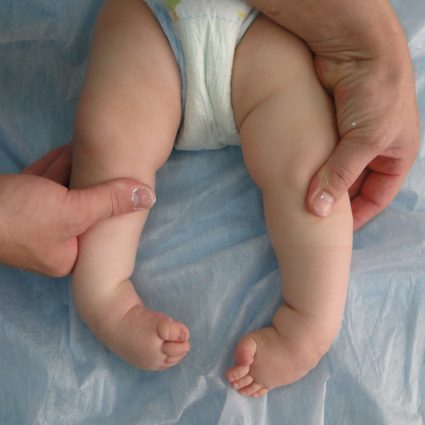
Для синдрома Гурлера характерна специфическая походка – на цыпочках с полусогнутыми ногами. Такая особенность возникает из-за ограничения движений по причине контрактур.
При физикальном обследовании выявляется следующее:
- живот увеличен, его передняя стенка у таких больных ослаблена, поэтому нередко могут развиваться пупочные и паховые грыжи;
- печень и селезенка увеличены;
- у мужчин часто выявляется гидроцеле – водянка яичка;
- в ряде случаев обнаруживаются нарушения координации движений;
- выявляется избыточный рост пушковых волос.
При инструментальном обследовании определяется следующее:
- на электрокардиограмме – признаки диффузного (распространенного) поражения сердечной мышцы;
- нарушения со стороны зрения и слуха;
- при офтальмоскопии обнаруживают помутнение и увеличение роговицы.
Наиболее характерными являются изменения, которые констатируют при проведении рентгенологического обследования:
- рентгенография позвоночника – позволяет выявить характерную деформацию позвоночного столба. При этом позвонки имеют форму куба, их края закруглены. Также отмечается кифоз – искривление позвоночника выпуклостью вперед;
- рентгенография грудной клетки – отмечается утолщение передних фрагментов ребер с одновременным истончением их задних отделов, укорочением и деформацией. Ребра при этом приобретают специфический вид и стают похожими на лопату или саблю. Также отмечается уменьшение и некоторое смещение головок плечевых костей;
- рентгенография таза – на ней выявляется сужение тазового кольца, вертлужные впадины (те, в которые являются «основой» формирования тазобедренного сустава) скошены, головки бедренных костей уменьшены;
- рентгенография трубчатых костей – отмечается истончение кортикального (поверхностного) слоя костей и некоторое расширение костномозгового канала;
- рентгенография костей кисти – помогает обнаружить недоразвитие дистальных (ногтевых) фаланг пальцев, укорочение и расширение средних и проксимальных (тех, которые находятся ближе к ладони) фаланг, а также пястных костей;
- рентгенография черепа – четко выявляется недоразвитость костей лицевого черепа, краниостеноз (сужение черепа в некоторых локациях) и макроцефалия (увеличение черепа).
У больных с диагнозом мукополисахаридоза типа IH выявляются следующие нарушения, которые могут расцениваться как отдельные болезни:
- пигментная дистрофия сетчатки – нарушение ее развития;
- глаукома – увеличение внутриглазного давления;
- застойные явления в области глазного дна;
- парезы – нарушение движения;
- параличи – полное нарушение двигательной активности в определенных участках опорно-двигательного аппарата.
Со временем у пациентов с диагнозом Гурлера развивается и необратимо прогрессирует умственная отсталость.
Мукополисахаридоз типа I-S
Другое название мукополисахаридоза типа I-S – болезнь Шейе. Заболевание было описано только в средине 20 столетия, хотя пациенты с характерными для него контрактурами пальцев кистей, нарушениями движений в крупных суставах и специфическими чертами лица встречались в практике клиницистов и раньше. Признаки могут комбинироваться с признаками синдрома Гурлера.
Этот вид мукополисахаридоза появляется в более позднем возрасте, чем при мукополисахаридозе типа IH, а его течение более благоприятное.
Характерной особенностью мукополисахаридоза типа I-S является то, что до 3-6 лет жизни развитие детей соответствует возрастной норме. Далее начинают проявляться следующие признаки:
- сгибательные контрактуры пальцев рук;
- ограничение разгибания в крупных и средних суставах – а именно локтевых, плечевых и лучезапястных.
Нарушение амплитуды движений со стороны нижних конечностей менее выраженное, чем при других видах описываемой патологии.
Полноценная клиническая картина этого вида мукополисахаридоза устанавливается к началу подросткового периода.
При физиологическом исследовании (осмотре) отмечается следующее:
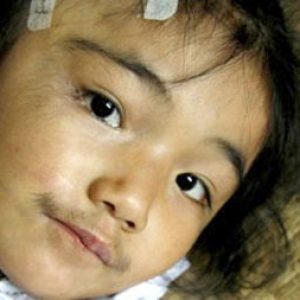
- черты лица довольно грубые;
- скелетная мускулатура развита хорошо;
- отмечается гипертрихоз (усиленное оволосение);
- кожные покровы на пальцах кистей (чаще) и стоп (реже) утолщены и натянуты;
- размеры печени и селезенки, как правило, не изменены.
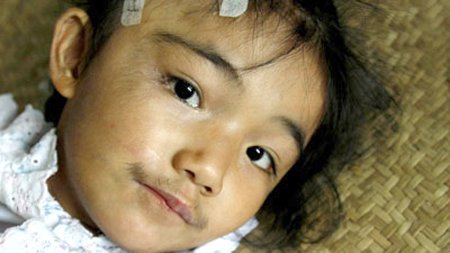
Помимо того, у таких пациентов определяются нарушения, которые могут расцениваться как отдельные заболевания:
- паховые или пупочные грыжи;
- синдром запястного канала – он развивается из-за выраженного сдавливания срединного нерва. Проявляется атрофией (недоразвитием) мышц тенара (возвышения в области большого пальца кисти) и парестезиями (нарушением чувствительности) в области 3, 4 и 5 пальцев;
- аортальный стеноз – сужение аорты;
- недостаточность клапанов аорты;
- пигментная дистрофия сетчатки;
- глаукома;
- помутнение роговицы.
Интеллект пациентов с диагнозом мукополисахаридоза типа I-S остается в норме, в отличие от больных с мукополисахаридозом типа IH.
Из инструментальных методов наиболее информативной является рентгенография. При этом определяются изменения, которые похожи на изменения при мукополисахаридозе типа IH, но они менее выражены.
Мукополисахаридоз типа II
Эта разновидность мукополисахаридоза еще называется болезнью Хантера. Патологией чаще всего страдают мальчики в возрасте 2-3 лет.
По клиническим проявлениям заболевание похоже на мукополисахаридоз типа IH – при этом наблюдаются:
- скароцефалия;
- грубые черты лица;
- низкий тембр голоса;
- затруднение дыхания – появляется из-за деформации костей лицевого скелета.
При дальнейшем прогрессировании заболевания присоединяются следующие признаки:
- снижается интеллект;
- нарушается координация движений;
- настроение у такого пациента неустойчивое – отмечается так называемая эмоциональная лабильность – от приступов смеха до плача. Такие резкие перепады настроения могут появиться в любое время.
При прогрессировании мукополисахаридоза типа II появляется и нарастает беспричинная агрессивность.
В отличие от мукополисахаридоза типа IH, нарушения со стороны позвоночника в виде кифоза не являются характерными, симптом «кошачьей спины» не отмечается.
При физикальном обследовании определяется следующее:
- выявляется незначительное увеличение селезенки и печени;
- на коже спины определяются специфические узелки.
При инструментальном обследовании выявляются:
- помутнение роговицы, которое прогрессирует;
- нарастающая тугоухость.
Из всех инструментальных методов диагностики наиболее информативной является рентгенография различных фрагментов опорно-двигательного аппарата – черепа, рудной клетки, таза и так далее. При этом рентгенологические признаки те же, что и при мукополисахаридозе типа I-S.
Пациенты с таким диагнозом склонны к развитию заболеваний дыхательной системы:
Следует учесть, что мукополисахаридоз типа II может развиваться в виде двух вариантов:
- неблагоприятного (вариант А);
- благоприятного (вариант В).
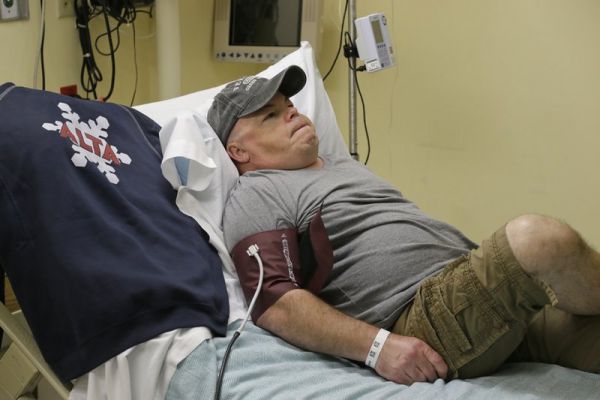
Симптоматика при благоприятном варианте невыраженная, выше описанные нарушения могут сопровождаться незначительной умственной отсталостью. Такие пациенты, как правило, доживают до 30 лет, в ряде случаев – и больше.
Мукополисахаридоз типа III
Мукополисахаридоз типа III описал американский педиатр Санфилиппо, поэтому патология получила еще одно название – болезнь Санфилиппо.
Заболевание диагностируется у 1 ребенка на 100-200 тысяч новорожденных.
Дети с мукополисахаридозом типа III в первые годы своей жизни развиваются согласно возрастным нормам, при этом могут наблюдаться отдельные проявления патологии – в частности, затруднение глотания и неуклюжая походка.
Начиная с 3-5 лет, ребенок начинает отставать от своих сверстников в физическом развитии, при этом он становится апатичным, безучастным к людям и событиям. Типичными являются следующие признаки:
- нарушение речи;
- огрубение черт лица;
- регулярное недержание мочи (не только ночное) и кала.
По мере взросления ребенка умственная отсталость нарастает и в конечном результате становится основным признаком этой разновидности мукополисахаридоза.
При физикальном обследовании обнаруживаются:
- несущественное увеличение печени и селезенки;
- равномерное усиление оволосения;
- контрактуры;
- задержка роста.

Результаты рентгенологического исследования при описываемой патологии или без патологических изменений, или же похожи на те, которые выявляются при мукополисахаридозе типа I-S, но менее выражены.
Прогноз при данном заболевании менее благоприятный, чем при других типах мукополисахарилоза – такие пациенты погибают в возрасте от 10 до 20 лет, причиной летального исхода становятся инфекционные осложнения.
Мукополисахаридоз типа IV
Мукополисахаридоз типа IV еще называется болезнью Моркио (по имени уругвайского педиатра, который ее описал). Патология встречается чаще, чем другие типы мукополисахаридоза – а именно у 1 ребенка на 40 тысяч новорожденных.
Дети с таким заболеванием до 1-3 лет развиваются без особенностей. Но далее появляются следующие нарушения:
- ребенок существенно отстает в росте от сверстников;
- в процессе развития у него отмечаются укорочение шеи и туловища;
- развиваются различные контрактуры;
- утолщается кожа;
- сила мышц уменьшается;
- слух ухудшается;
- черты лица становятся грубыми;
- возникают различные деформации грудной клетки.
Также при мукополисахаридозе типа IV отмечаются нарушения, которые могут расцениваться как отдельные патологии:
- вальгусная деформация конечностей – искривление в направлении наружу;
- сколиоз (искривление позвоночника в боковой плоскости) или кифоз;
- паховая грыжа;
- пупочная грыжа;
- дистрофия роговицы.
Интеллект при таком виде мукополисахаридоза сохранен.
Диагноз описываемой патологии подтверждают с помощью рентгенологического исследования, оно в данном случае наиболее информативное. Это следующие его виды:
- рентгенография позвоночника – на снимках определяются искривления позвоночного столба в виде кифоза и сколиоза, а также расширение и уплощение тел позвонков;
- рентгенография таза – выявляются множественные деформации тазового костного кольца, неровность его контуров;
- рентгенография конечностей – выявляются уплощение головок бедренных костей, выразительное укорочение костей предплечья и деформация костей стоп.
Больные с мукополисахаридозом типа IV живут, как правило, менее 20 лет. Летальный исход наступает на фоне ряда сопутствующих заболеваний, которые в конечном результате осложняются сердечно-легочной недостаточностью.
Мукополисахаридоз типа VI
Другое название мукополисахаридоза типа VI – болезнь Марото-Лами (по именам двух французских врачей, описавших эту патологию).
Клинические признаки во многом «перекликаются» с симптоматикой других видов мукополисахаридоза, но наиболее выраженными являются:
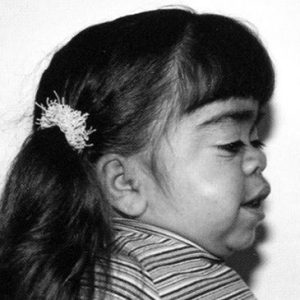
- заметное отставание в росте;
- укорочение шеи – иногда кажется, что голова пациента «сидит» чуть ли не на его плечевом поясе;
- бочкообразная деформация грудной клетки.
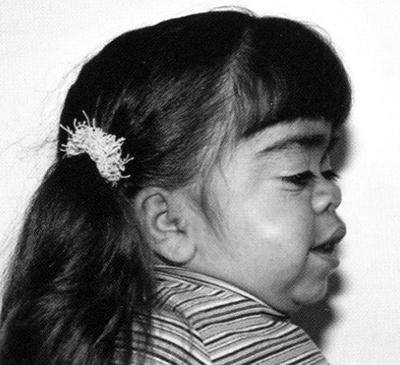
У таких больных очень часто возникают простуды – данный признак позволяет заподозрить именно эту, а не другую форму мукополисахаридоза.
При физикальном обследовании выявляется следующее:
- печень и селезенка зачастую увеличены;
- могут диагностироваться паховые грыжи.
В интеллектуальном плане ребенок развивается без нарушений – у него сохранена способность мыслить и решать интеллектуальные задачи.
Различают два варианта такого вида мукополисахаридоза:
- классический;
- мукополисахаридоз со несущественной выраженностью клинических проявлений.
Подтвердить диагноз поможет рентгенологическое обследование – на снимках определяются:
- деформация позвонков – они становятся кубовидными или клиновидными;
- треугольная деформация тазового кольца;
- гипоплазия (недоразвитие) и деформация головок бедренных костей;
- укорочение малоберцовых костей.
Мукополисахаридозы типа VII и VIII

Мукополисахаридоз типа VII клинически протекает, как болезнь типа III, разница между ними обнаруживается только в результатах биохимических исследований.
Мукополисахаридоз типа VIII по симптоматике похож на мукополисахаридоз типа IV. Но отличие в проявлениях имеется – это нарушение интеллектуального развития, которое не наблюдается при мукополисахаридозе типа IV.
Диагностика
При постановке диагноза мукополисахаридоза основываются на жалобах, анамнестических данных, результатах дополнительных методов обследования – физикальных, инструментальных, лабораторных. Заподозрить наличие мукополисахаридоза можно по характерным деформациям скелета, которые «бросаются» в глаза, но определить тип описываемого заболевания представляется возможным только с помощью дополнительных методов обследования. Главными из таких методов являются:
- рентгенография;
- выявление гликозаминогликанов в моче;
- определение активности ферментов в клеточных культурах.
При клинических признаках нарушений со стороны внутренних органов привлекаются методы их обследования:
- электрокардиография;
- ультразвуковое исследование печени и селезенки;
- электроэнцефалография;
- биохимический анализ крови
и многие другие.
Дифференциальная диагностика
Дифференциальную (отличительную) диагностику проводят между различными типами мукополисахаридозов.
Осложнения
Наиболее частыми осложнениями мукополисахаридозов являются:
- контрактуры;
- сердечная недостаточность;
- легочная недостаточность.
Лечение
Терапия мукополисахаридоза, с помощью которой можно бы устранить болезнь, а не только ее проявления, не разработана. Таким пациентам проводят симптоматическое лечение – оно может быть:
- консервативное – переливание крови, гормонотерапия, витаминотерапия и так далее;
- оперативное – эндопротезирование головок бедренных костей, ликвидация грыж, хирургическое вмешательство для устранения контрактур и исправления деформаций скелета и другие.
Важными являются:
- лечение инфекций дыхательных путей;
- коррекция нарушений зрения и слуха.
Профилактика
Так как патология возникает из-за генетических нарушений, специфических методов профилактики не существует. Но риск генных нарушений, которые способны привести к развитию различных типов мукополисахаридоза, можно снизить, если обеспечить женщине нормальные условия протекания беременности. Это:
- отказ от вредных привычек;
- соблюдение режима работы, отдыха, сна;
- оптимистический настрой.
Прогноз
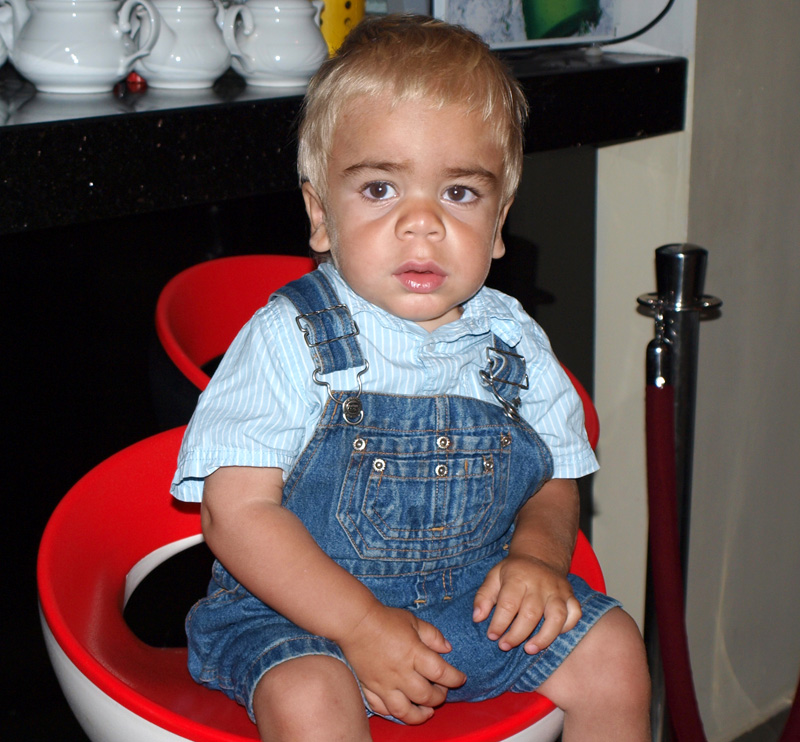
Прогноз при мукополисахаридозе в целом неблагоприятный, это касается всех типов описываемого заболевания. В процессе жизни и развития в тканях прогрессивно накапливаются продукты обмена, которые возникают из-за отсутствия тканевых ферментов. Это приводит к патологическим изменениям со стороны практически всех органов и систем. Если патология запущена, то летальный исход может наступить не только из-за сердечно-легочной, но и полисистемной недостаточности.
Консервативная терапия выполняет роль поддерживающей – применение любых медикаментозных средств ведет к временному улучшению, но не способно компенсировать нехватку тканевых ферментов.
Ковтонюк Оксана Владимировна, медицинский обозреватель, хирург, врач-консультант
7,954 total views, 1 views today

 (45 голос., средний: 4,31 из 5)
(45 голос., средний: 4,31 из 5)










